Functional annotation#
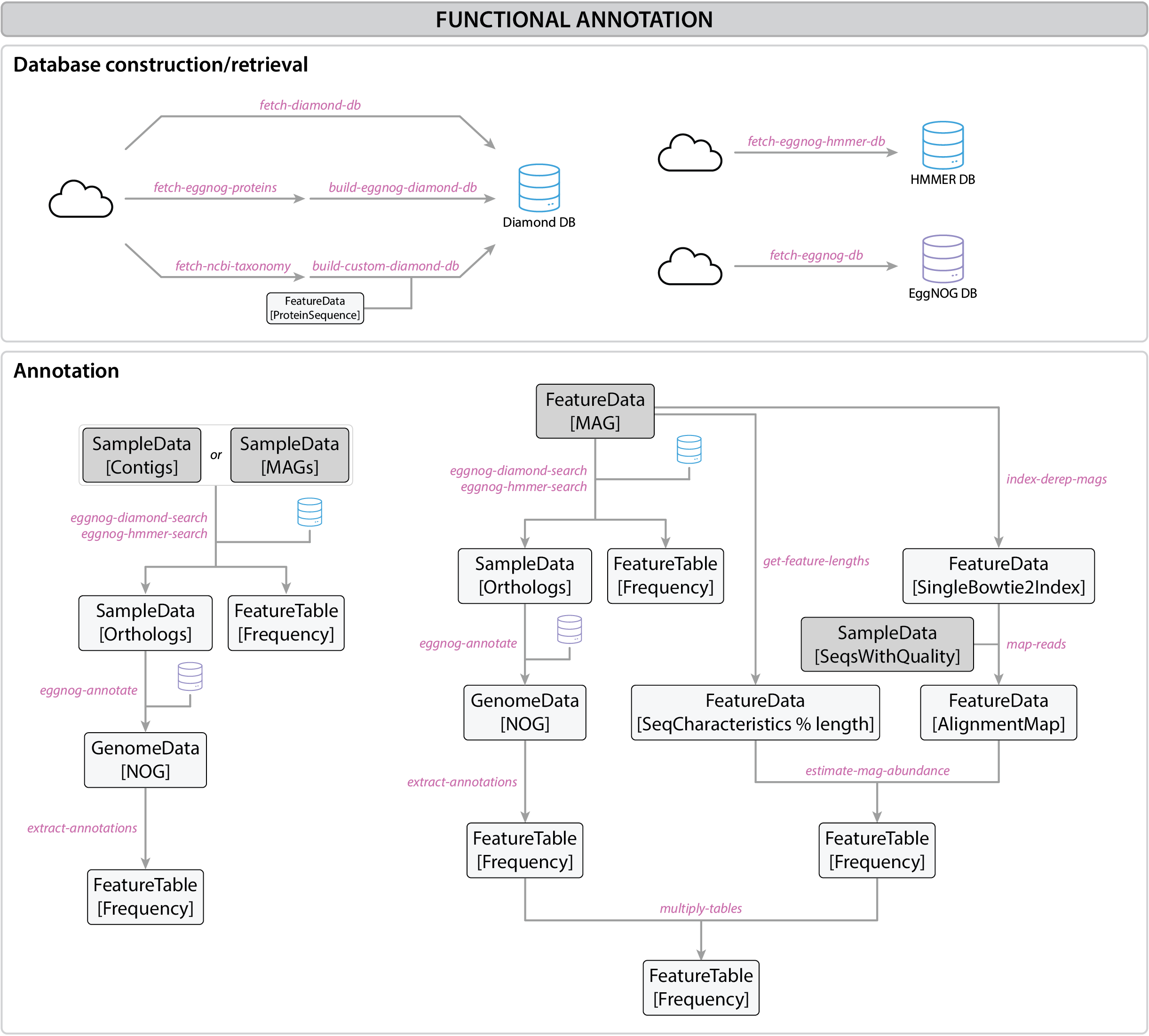
Fig. 3 Functional annotation workflow#
Functional annotation of metagenome-assembled genomes (MAGs) involves the identification and classification of genes within these reconstructed genomes to understand their roles and potential functions. MAGs are recovered from DNA directly extracted from complex microbial communities, bypassing the need to culture the organisms.
This process provides insights into the genes that code for enzymes, transporters, and other proteins critical to the survival and function of the microbes in various ecosystems. Annotating these genomes allows for the study of their contributions to nutrient cycles, disease processes, or specialized ecological functions, to name only a few examples.
This workflow outlines the step-by-step process for functional annotation of MAGs or contigs using tools like EggNOG and the Diamond aligner in QIIME2.
Note
Functional annotation can be performed on fully reconstructed MAGs or directly on contigs (the contiguous sequences assembled from sequencing reads). Annotating contigs can provide early insights into important functional genes even before complete genomes are assembled. Annotating MAGs has the added benefit of seeing how these annotated genes are connected and organized in a single genome.
In this tutorial, we will focus on functional annotation of our previously reconstructed MAGs (see Recovery of MAGs section)
Warning
Functional annotation can be highly resource-intensive. Ensure that your system has sufficient CPU and memory resources before running these commands.
For more information on the tools used in this workflow, refer to their official documentation:
EggNOG-mapper: eggnogdb/eggnog-mapper
DIAMOND: bbuchfink/diamond
QIIME 2: qiime2