Taxonomic classification#
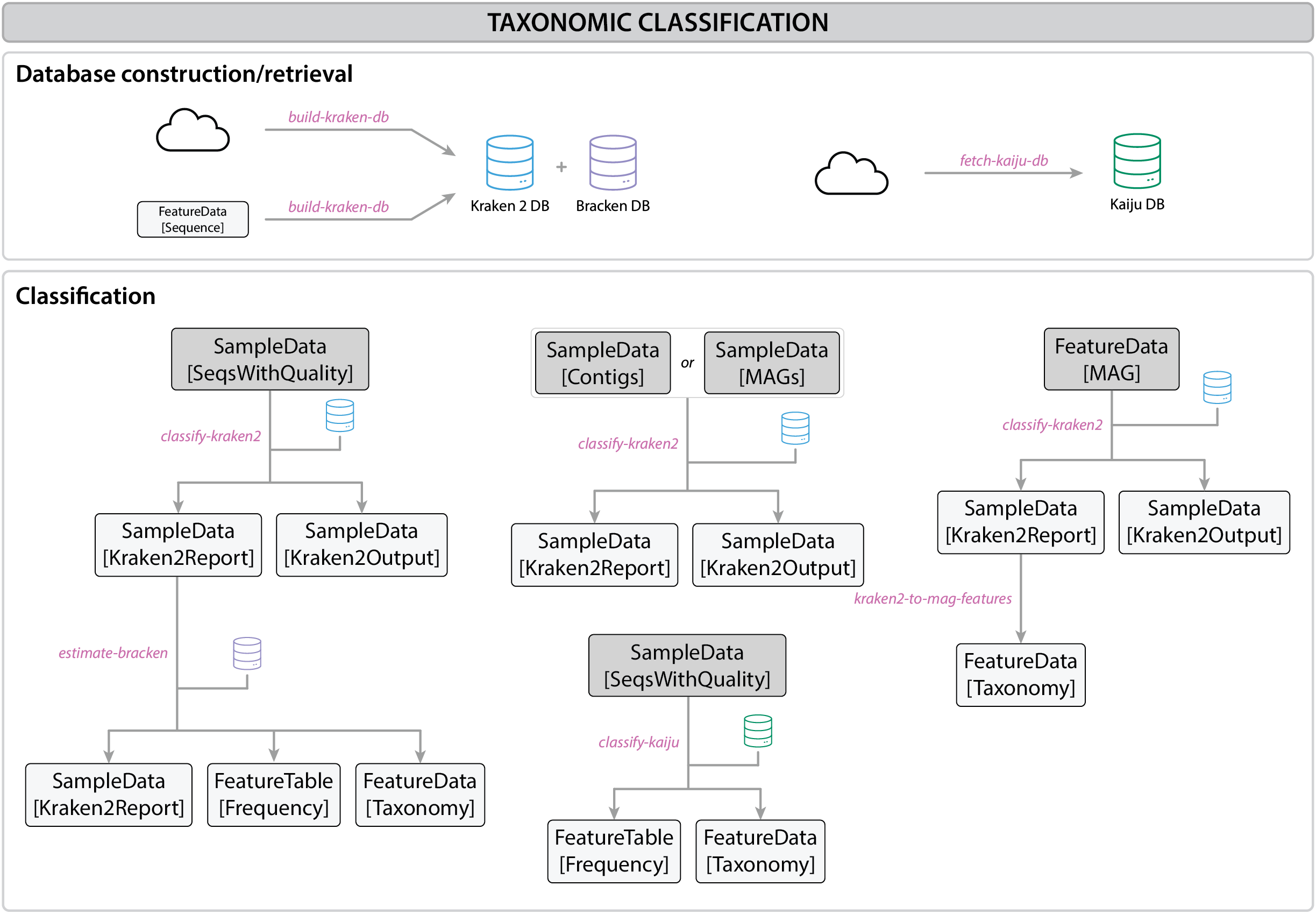
Fig. 2 Taxonomic classification workflow#
Read-based classification overview#
Read-based classification is commonly used to determine the taxonomic groups present within a given sample. This technique is useful for assessing the biodiversity or the composition of microbial communities by assigning DNA reads to known organisms. One significant advantage of read-based classification is that it allows for the classification of all reads, including those that may not be directly involved in downstream analyses (e.g. assemblies or MAGs).
Key Factors Influencing Read Classification: The outcome of read classification is heavily influenced by the selection of reference databases. These databases vary in size, quality, and scope, which means that the more comprehensive and accurate the reference database is, the more accurate the classification of reads will be. Some databases might be specific to certain taxonomic groups, while others could provide a broader reference, potentially affecting the results depending on the sample type and research goals.
See also
For more information and a benchmark, consult Ye et al., 2019.
Kraken 2: DNA-to-DNA classification#
Kraken 2 is a DNA-to-DNA classification tool that assigns taxonomic labels to reads by directly comparing k-mers (short DNA sequence fragments of a fixed length, typically 31 base pairs) from the query read to a database of known sequences. Kraken 2 classifies the read based on the majority of k-mer matches within the read, providing fast and accurate taxonomic classification.
See also
For more information on Kraken 2, consult Wood et al., 2019.
Kaiju: protein-based classification#
Kaiju compares reads by translating DNA sequences into protein sequences (similar to BLASTx). This allows Kaiju to identify organisms accurately when nucleotide sequences are too divergent to be identified with DNA-based methods. Kaiju uses a fast exact matching algorithm based on Burrows-Wheeler Transform (BWT) and FM-index to align translated DNA reads against a reference database of protein sequences.
See also
For more information on Kaiju, consult Menzel et al., 2016.
Warning
Taxonomic classification can be highly resource-intensive. Ensure that your system has sufficient CPU and memory resources before running these commands.
For more information on the tools used in this workflow, refer to their official documentation:
Kraken 2: DerrickWood/kraken2
Kaiju: bioinformatics-centre/kaiju